- info@i-genie.co.uk
- 0207 148 4785
Proteins are essential for various cellular functions, providing vital amino acids for humans. Understanding proteins is crucial for human biology and health, requiring advanced machine-learning models for protein representation. Self-supervised pre-training, inspired by natural language processing, has significantly improved protein sequence representation. However, existing models need help handling longer sequences and maintaining contextual understanding. Strategies like linearized and sparse approximations have been used to address computational demands but often compromise expressivity. Despite advancements, models with over 100 million parameters struggle with larger inputs. The role of individual amino acids poses a unique challenge, requiring a nuanced approach for accurate modeling.
Researchers from the Tokyo Institute of Technology, Japan, have developed ProtHyena. This rapid and resource-efficient foundation model incorporates the Hyena operator for analyzing protein data. Unlike traditional attention-based methods, ProtHyena is designed to capture both long-range context and single amino acid resolution in real protein sequences. The researchers pretrained the model using the Pfam dataset. They fine-tuned it for various protein-related tasks, achieving performance comparable to or even surpassing state-of-the-art approaches in some cases.
Traditional language models based on the Transformer and BERT architectures demonstrate effectiveness in various applications. Still, they are limited by the quadratic computational complexity of the attention mechanism, which restricts their efficiency and the length of context they can process. Various methods have been developed to address the high computational cost of self-attention for long sequences, such as factorized self-attention used in sparse Transformers and the Performer, which decomposes the self-attention matrix. These methods allow for processing longer sequences but often come with a trade-off in model expressivity.
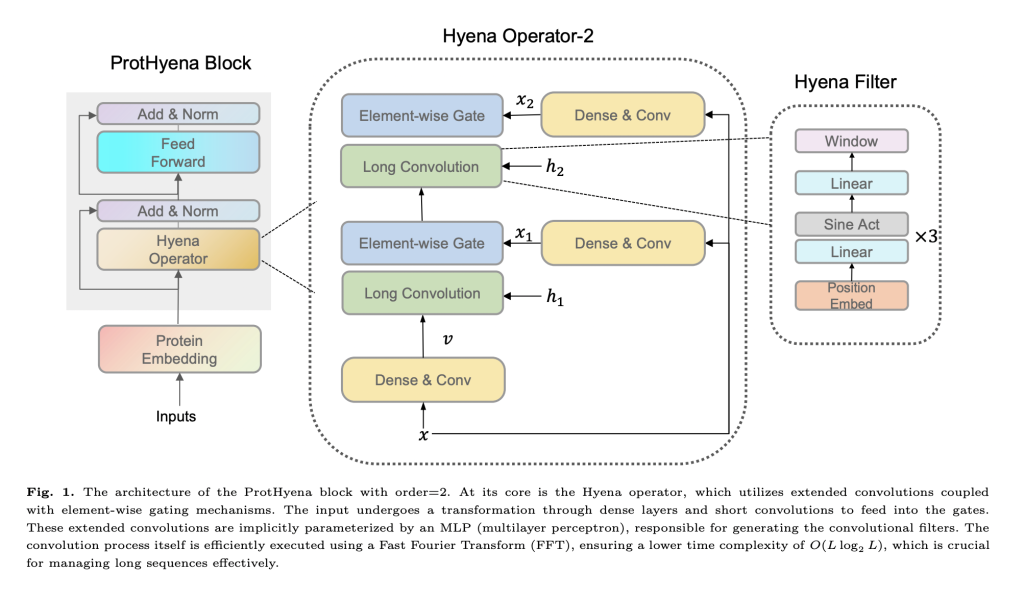
ProtHyena is an approach that leverages the Hyena operator to address the limitations of attention mechanisms in traditional language models. ProtHyena uses the natural protein vocabulary, treating each amino acid as an individual token, and incorporates special character tokens for padding, separation, and unknown characters. The Hyena operator is defined by a recurrent structure comprising long convolutions and element-wise gating. The study also compares ProtHyena with a variant model called ProtHyena-bpe, which employs byte pair encoding (BPE) for data compression and utilizes a larger vocabulary size.
ProtHyena addresses the limitations of traditional models based on the Transformer and BERT architectures. ProtHyena achieved state-of-the-art results in various downstream tasks, including Remote Homology and Fluorescence prediction, outperforming contemporary models like TAPE Transformer and SPRoBERTa. Regarding Remote Homology, ProtHyena reached the highest accuracy of 0.317, surpassing other models that scored 0.210 and 0.230. For Fluorescence prediction, ProtHyena demonstrated robustness with a Spearman’s r of 0.678, showcasing its ability to capture complex protein properties. ProtHyena also showed promising results in Secondary Structure Prediction (SSP) and Stability tasks, although the provided sources did not mention specific metrics.
In conclusion, ProtHyena, a protein language model, integrates the Hyena operator to address the computational challenges faced by attention-based models. ProtHyena efficiently processes long protein sequences and delivers state-of-the-art performance in various downstream tasks, surpassing traditional models with only a fraction of the parameters required. The comprehensive pre-training and fine-tuning of ProtHyena on the expansive Pfam dataset across ten different tasks demonstrate its ability to capture complex biological information accurately and accurately. Adopting the Hyena operator enables ProtHyena to perform at a subquadratic time complexity, offering a significant leap forward in protein sequence analysis.
Check out the Paper. All credit for this research goes to the researchers of this project. Also, don’t forget to follow us on Twitter. Join our 36k+ ML SubReddit, 41k+ Facebook Community, Discord Channel, and LinkedIn Group.
If you like our work, you will love our newsletter..
Don’t Forget to join our Telegram Channel
The post Researchers from the Tokyo Institute of Technology Introduce ProtHyena: A Fast and Efficient Foundation Protein Language Model at Single Amino Acid Resolution appeared first on MarkTechPost.